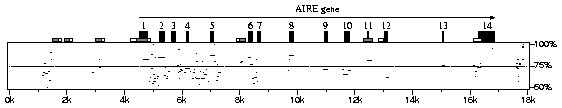
PipMaker computes alignments of similar regions in two DNA sequences. The resulting alignments are summarized with a ``percent identity plot'', or ``pip'' for short.
To generate a pip, such as the one shown above, PipMaker requires four user-supplied files. The first sequence file is depicted along the horizontal axis. Interspersed repeats in the first sequence are indicated by various kinds of triangles, whose locations are supplied by a mask file of the first sequence. (The user generates this file using the RepeatMasker program, available on the web at the Institute for Systems Biology.) A file of gene and exon positions allows PipMaker to draw the locations of exons and indicate the directionality of genes, shown as black boxes and long arrows, respectively. Finally, the user provides a second sequence file. CpG islands in the first sequence are independently determined by PipMaker and are shown as low boxes.
PipMaker compares the first and second sequences. Alignments are plotted according to the position in the first sequence file. The light horizontal line through the middle of the plot indicates 75% nucleotide identity. This version of PipMaker compares the first sequence with both the second sequence and its reverse complement, so matching regions need not occur in the same orientations and relative positions in the two sequences. (Advanced PipMaker can optionally enforce the condition that matching regions appear in the same relative order and/or in the same orientation. To test PipMaker, copy the four provided files to your computer and submit them to PipMaker to generate the above sample pip.
PipMaker processes the contents of the following four files. For each of those, you can either paste the data into the multi-line textarea or, if your browser supports it, give the filename in the subsequent single-line field.
>exactly one header line, rest contain ACGT ONLY ACGTACGTACGT CGTACGTACGTA GTACGTACGTAC TACGTACGTACG
413 5.6 0.0 0.0 HUMAN 1 54 (92195) C Alu SINE/Alu (238) 62 9 SINE/Alu (238) 62 9In certain circumstances, RepeatMasker produces some summary statistics that precede lines of that form -- those additional lines are ignored by PipMaker. Inclusion of this file is optional, but it is strongly recommended for mammalian sequences, since otherwise biologically insignificant matches due to repeats will be computed. If this file is omitted, then the pip will not include icons showing the positions of interspersed repeat elements.
My favorite genomic region > 100 800 Gene 1 100 200 300 400 600 800 < 1000 2000 Second Gene + 1100 1900 1000 1200 1800 2000 ...If the exon numbers supplied by PipMaker are not adequate, a number (or a name, for that matter) can be specified at the end of exon-position lines.
The following three files are returned as attachments to an email message. You will need a MIME-aware email program to read them.
... 2870-2926 <--> 2281-2337 68% (57 nt) 3117-3128 <--> 2500-2511 83% (12 nt) 3129-3179 <--> 2513-2563 73% (51 nt) ...The first line asserts that positions 2870-2926 in the first sequence (57 basepairs) aligns to positions 2281-2337 of the second sequence, without gaps and at 68% identity.
0 . : . : . : . : . :
2870 GCCCAGGCCTGGGCAGCGAGAGGGCCCTGCTCCCCGCTCAAGGCTCCCAG
||||||:| ||::||::|:|||| ||||||||:||:|||| ::|::::||
2281 GCCCAGACATGAACAATGGGAGGCCCCTGCTCTCCACTCACAACCTTTAG
50 .
2920 GACATTC
:||||||
2331 AACATTC
The pip consists of rows that show sequence conservation and features along segments of the first sequence. Each short horizontal line inside the large box corresponds to a section of an alignment bounded by successive gaps (or an end of the alignment). For instance, suppose that one of the alignments computed for your two sequences begins as follows.
0 . : . : . : . : . :
19163 GCGGCTCCATGTCACCTGCGGGCAAGGGGCTGGTGTGGAAAGCCCCACGG
||:||| || ::|||||||:||:: ||:-||| :::||||| |||||||
6465 GCAGCTACAGACCACCTGCAGGTGTGGA CTGTCACGGAAACCCCCACGT
50 . : . : . : . : . :
19213 CATGGTGGAAAGTCCGAAATTCTACAGGGGCCTCTTTGTTAAACCTC
-||:||||||||||:||||||||||||:|:||:|:||||:| |::|---
6514 G TGATGGAAAGTCCAAAATTCTACAGGAGTCTTTCTGTTGATCTCCAGT
...
In this portion of the alignment, there are three gap-free pieces.
The first covers positions 19163-19190 of the first sequence at 64%
nucleotide identity, the second spans 19192-19213 at 68% identity, and the
third covers 19215-19259 at 78% identity.
This would be depicted in the pip by three horizontal line segments that
indicate the positions in the first sequence and the percent identity.
Icons along the top of the box have the following meanings.