Advanced PipMaker processes the contents of the same four files as does the basic form of PipMaker. The pip begins with a one-line overview of the first sequence. If an underlay file was given, the first row of the overview shows it. The bottom (and perhaps only) row of the overview shows aligned regions in green and strongly aligned regions (at least 100 bp without a gap and with at least 70% nucleotide identity) in red. Legends for the icons representing interspersed repeats and the meanings of colors in the underlay and annotation files (see below) are place on separate pages. In addition, a number of choices, options, and new capabilities are provided.
TABLE OF CONTENTS
Regions of interest in the first sequence, such as the locations of exons or regulatory elements, can be colored by vertical stripes. To use this feature of Advanced PipMaker, upload a first sequence underlay file, which should begin with lines like:
Red Strongly_conservedfollowed by lines like:
1000 2000 Strongly_conservedThe first set of lines describes the intended interpretion of each color, while the second tells where the colored stripes are to be placed. In color descriptions, colors (the first word on each line) must be selected from a restricted list. The second word on each of these lines is chosen arbitrarily by the user. It is possible to paint just the upper half of a vertical stripe by requesting, e.g.:
50060 50227 Strongly_conserved +which was done for the BTK pip. Use "-" to paint just the lower half. This permits annotations to differentiate between the two strands, or to plot potentially overlapping features like gene predictions and database matches. (Painting over a stripe, as with the EST region in
1200 1500 EST 1000 2000 Strongly_conservedwill make it invisible.) Beware that the appearance of the colors will vary from one printer or monitor to the next.
The user-supplied file (example) defines various types of hyperlinks and associates a color with each of them, then specifies the type, position, description, and URL for each annotated feature. For instance, in the example,
%define type %name PubMed %color Bluerequests that each feature identified as a PubMed entry be colored blue. The name must be a single word, perhaps containing underline characters, as in Entry_in_GenBank. Colors start with capital letters. The subsequent entry
%define annotation %type PubMed %range 1 2000 %label Yang et al. 1997. Daxx, a novel Fas-binding protein... %summary Yang, X., Khosravi-Far, R. Chang, H., and Baltimore, D. (1997). Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89(7):1067-76. Click to see Abstract. %url http://www.ncbi.nlm.nih.gov:80/entrez/ query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9215629&dopt=Abstractassociates a PubMed annotation with positions 1-2000 in the first sequence. Note that summaries and URLs (but not labels) can be broken into several lines for convenience; the line breaks are removed when the file is read, but they are not replaced with spaces. Thus a continuation line for a summary typically begins with a space to separate it from the last word of the previous line, while a URL continuation does not. If the summary is omitted, it is assumed to be the same as the label. Annotations can overlap, as shown in this example. Each %define line except for the first one should be immediately preceded by a blank line.
Clicking on this blue bar will bring up a page that displays a description of the feature (as given in the %summary stanza), including a "hand" icon that can be clicked on to visit the specified URL. (Within the PDF document, the Acrobat Reader's navigational controls should be used; e.g., the web browser's Back button will not return to the PIP.)
The alignment program can optionally report only regions of similarity between the first sequence and the second sequence in its given orientation. The default is to align the first sequence with both the second sequence and the reverse complement of the second sequence.
With the default setting of "Show all matches", it is possible for one region of the first sequence to align with several regions of the second sequence because of duplications of a gene or an exon, or because of incomplete masking of interspersed repeats or low-complexity regions. Such duplications cause lines to appear one over the other in the pip. PipMaker provides two options for eliminating such duplicate matches, each with its own strengths and weaknesses.
If the "Chaining" option is choosen, then PipMaker will identify and plot only matches that appear in the same relative order in the first and second sequences. This option should be used only if the genomic structures of the two sequences are known to be conserved, since otherwise a duplication might avoid detection. With Chaining, the alignment program is run with lower thresholds (i.e., higher sensitivity).
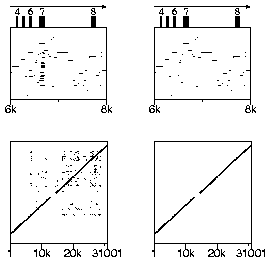
For an example of chaining, consider the first pip shown below, which is taken from a larger pip based on a 31 kb sequence. As the pip indicates, exon 7 of the first sequence has a number of matches in the second sequence, presumably due to duplications of that exon or of the entire gene. The dotplot view of the entire alignment, shown at the left of the second row, also indicates the duplication (at around position 7000 on the horizontal axis), as well as duplications in later exons. The panels on the right show the results of specifying the "Chaining" option.
An alternative method for avoiding duplicate matches is provided by the "Single coverage" option, which selects a highest-scoring set of alignments such that any position in the first sequence can appear in at most one alignment (though there is no guarantee that order of matching regions is identical in the two sequences). The following three dotplots show a case where this option works better than does chaining. The top panel shows all matches in a gene cluster where the first sequence has six copies of the gene, while the second sequence has four. With chaining, only four of the genes can be matched, as shown in the second panel. The "Single coverage" option selects one match for each region of the first sequence (panel 3).

Further discussion of this example can be found on the Examples page, under beta-globin.
Chaining is preferable to the "Single coverage" option in cases where (1) the second sequence is contiguous, (2) the comparison is with just a single strand of the second sequence and (3) the order of conserved regions is identical in the two sequences, since under conditions 1-3 the results from the "Chaining" option will be more biologically meaningful. (Also, the comparison will be faster.) However, the three-panel example shows that if condition (3) does not hold, the "Chaining" option may give inferior results. A strength of the "Single coverage" option is that it guarantees single coverage even if the second sequence is compared in both orientations, or if it is fragmented (see below). In such cases, the "Chaining" option is applied separately each time the first sequence is compared with an orientation or fragment of the second sequence, so multiple coverage can result.
The default settings of PipMaker are tuned to perform well when comparing two mammalian sequences. The option for high sensitivity works better for a sequence pair at a greater evolutionary distance, such as human-fugu. However, if two rather similar sequences are compared with this setting (e.g. human-mouse), the alignment program can run much longer than desired, so we have set the server to terminate execution after a few minutes. This is long enough to permit, say, a successful human-fugu alignment over two BACs.
The user selects output files from the following list: the pip, a one-page dotplot view of the alignments, the condensed form of the alignment, the traditional textual form of the alignment, an analysis of the exons, raw blastz output, and files predicting the order and orientation of the contigs (assuming the second sequence file is broken into contigs). The last three forms of output are described in more detail below.
The user can supply an optional title for the pip. (By default, the title is taken from the first line of the exons file.) The user can request that the output be in PostScript format instead of PDF format.
For the optional analysis of exons, a program attempts to use the alignments to map each position in the "exons" file into the corresponding position in the second sequence. For each coding-region specification (i.e., line beginning with "+") the first and last codons are displayed, and for each intron the first and last two nucleotides are shown. These tri- and dinucleotides are shown for the first sequence and, if possible, for the second sequence. If the coding region is specified, then the putative coding region is printed (for both sequences, if possible). For example, if the exons file contains:
< 27591 30475 L44L + 27641 30438 27591 27661 27932 28054 29660 29727 29918 30023 30436 30475then the program might generate:
< L44L: 27591-30475 35159-37479 CDS: 27641-30438 35202-37442 TTA-CAT TTA-CAT 5: 27591-27661 35159-35222 CT-AC CT-AC 4: 27932-28054 35586-35708 CT-AC CT-AC 3: 29660-29727 36581-36648 CT-AC CT-AC 2: 29918-30023 36976-37081 CT-AC CT-AC 1: 30436-30475 37440-37479 >L44L, putative CDS for sequence 1 (321 bp) ATGGTTAACGTCCCTAAAACCCGCCGGACTTTCTGTAAGAAGTGTGGCAA GCACCAACCCCATAAAGTGACACAGTACAAGAAGGGCAAGGATTCTCTGT ....This output shows that the L44L coding region in both sequences begins with ATG (the reverse complement of CAT) and ends with TAA. Similarly all four introns conform to the normal GT-AG splicing consensus.
When the second sequence file is split into contigs, the output is somewhat more complicated. It begins with an enumeration of the contigs (using their FastA header lines), and positions in the second sequence are identified as i:j which denotes position j in contig i. For instance, the index might include:
Index of fragments of the second sequence: 1: >Contig101 2: >Contig102and the listing of exon positions might contain:
< L44L: 27591-30475 1:35159-2:1479 CDS: 27641-30438 1:35202-2:1442 TTA-CAT TTA-CAT 5: 27591-27661 1:35159-1:35222 CT-AC CT-AC 4: 27932-28054 1:35586-1:35708 CT-AC CT-AC 3: 29660-29727 2:581-2:648 CT-AC CT-AC 2: 29918-30023 2:976-2:1081 CT-AC CT-AC 1: 30436-30475 2:1440-2:1479Here, the left-most CDS position (end of the stop codon) aligns with position 35202 in contig 1, which has FastA header line ">Contig101".
The alignment file produced by PipMaker's blastz program can be viewed in laj, which is an interactive tool for viewing and manipulating pairwise alignment output. The laj program, which is written in Java, must be down-loaded and run on the users computer. Also, alignments in this format can be submitted to the SGP-1 gene prediction program.
PipMaker can predict the order and orientation of contigs (contiguous segments) in the second sequence. This will return three files:
The second sequence is assumed to consist of contigs, separated either by FastA header lines, or by runs of 100 or more letters "N". PipMaker starts by replacing each such run of N's by a synthesized FastA header line of the form ">word.Ci", where ">word" is the first word on the closest preceding FastA header line present in the original submission, and i runs 2, 3, 4, ... . Thus, a second sequence consisting of two N-free segments separated by 100 Ns, such as
>GeneX an imaginary gene ACGT...TGCA NNNN...NNNN TGCA...ACGTbecomes:
>GeneX an imaginary gene ACGT...TGCA >GeneX.C2 TGCA...ACGT(In the preceding larger example, the second sequence began with the FastA line
>gi|9966970|gb|AC011189.5|AC011189 Homo sapiens chromosome 17 clone RP11-231G16 map 17, WORKING DRAFT SEQUENCE, 39 unordered piecesfrom which PipMaker extracted the word "AC011189".)
With the default setting, it is essential that interspersed repeats in the second sequence must not be replaced by long runs of N's in the file submitted to PipMaker. (The letter X could be used, but is not recommended.) On the other hand, the prediction will generally be much more reliable if positions of repeats in the first sequence are given to PipMaker, since otherwise the predictions may be based on spurious alignments.
Optionally, the user can disable the "Break at NN.." option, and thereby get order-and-orientation predictions when NN..N is used to mask regions of the second sequence. XX
Notes on configuring Acrobat Reader: Follow the menu File > Preferences > Weblink and set the Link Information selector to "Always Show". (You will need to run Acrobat Reader in stand-alone mode to do this, since the browser plug-in does not have its own File menu.)
With this option, when the Pip is viewed using Adobe Acrobat Reader, a message that corresponds to the mouse position appears, either in the status area at the bottom of the Acrobat window or as a tooltip. When the mouse is positioned in a region of the pip corresponding to a local alignment, the message describes the region covered by the alignment, whereas between two alignments it describes the gap. The first word on the FastA header line for the second sequence, or a contig thereof, is reported. This indicates which contig aligns with this region of the first sequence.
For example, the following messages might be associated with various regions of a certain sequence (submitted as the first sequence to PipMaker).
region message 1-1400 1400 bp unmatched 1401-1586 1401-1586 matches 3146-3324 of Contig35 1587-2191 605 bp gap; 889 bp in Contig35 2192-3367 2192-3367 matches 4214-5531 of Contig35 3368-3888 521 bp gap; between Contig35 and Contig41- 3889-3934 3889-3934 matches 6-51 of Contig41- 3935-4089 155 bp gap; 1013 bp in Contig41- 4090-4375 4090-4375 matches 1065-1340 of Contig41-The notation "Contig41-" refers to the reverse complement of the sequence with FastA header ">Contig41 ...". In this example, the first sequence has two local alignments with Contig35, followed by two local alignments to the reverse complement of Contig41. Note that the latter pair of alignments are separated by 155 bp (positions 3935-4089) in the first sequence and 1013 bp in Contig41-, perhaps due to an insertion at this point in the second sequence.
These messages in the pip are not intended to be clickable -- we are slightly abusing the URL mechanism by storing the messages as URLs, because it is the most convenient way to display a short string.
PipMaker attempts to eliminate spurious alignments caused by interspersed repeat elements and low-complexity regions, yet indicate meaningful alignments that may involve such elements. For instance, an interpersed repeat or tri-nucleotide repeat sequence may be part of a protein-coding region. The strategy is not perfect, however, and the user may want to take contol of the way these elements are handled during construction of the alignment.
The alignment program used by PipMaker interprets lower case letters as indicating regions of the sequence that are to be masked at early stages of the alignment process, but not at later stages. The alignment program follows the general design of the "gapped Blast" family of programs, which start by finding short, exact matches, then extend those matches to alignments that include gaps. PipMaker ignores regions of the first sequence that contain lower case letters when searching for exact matches, but utilizes those regions when expanding exact matches to form longer alignments. (Regions containing only "N" or "X" characters are not aligned in either phase.) Users of the Advanced PipMaker page can control masking by submitting sequences that follow this convention.